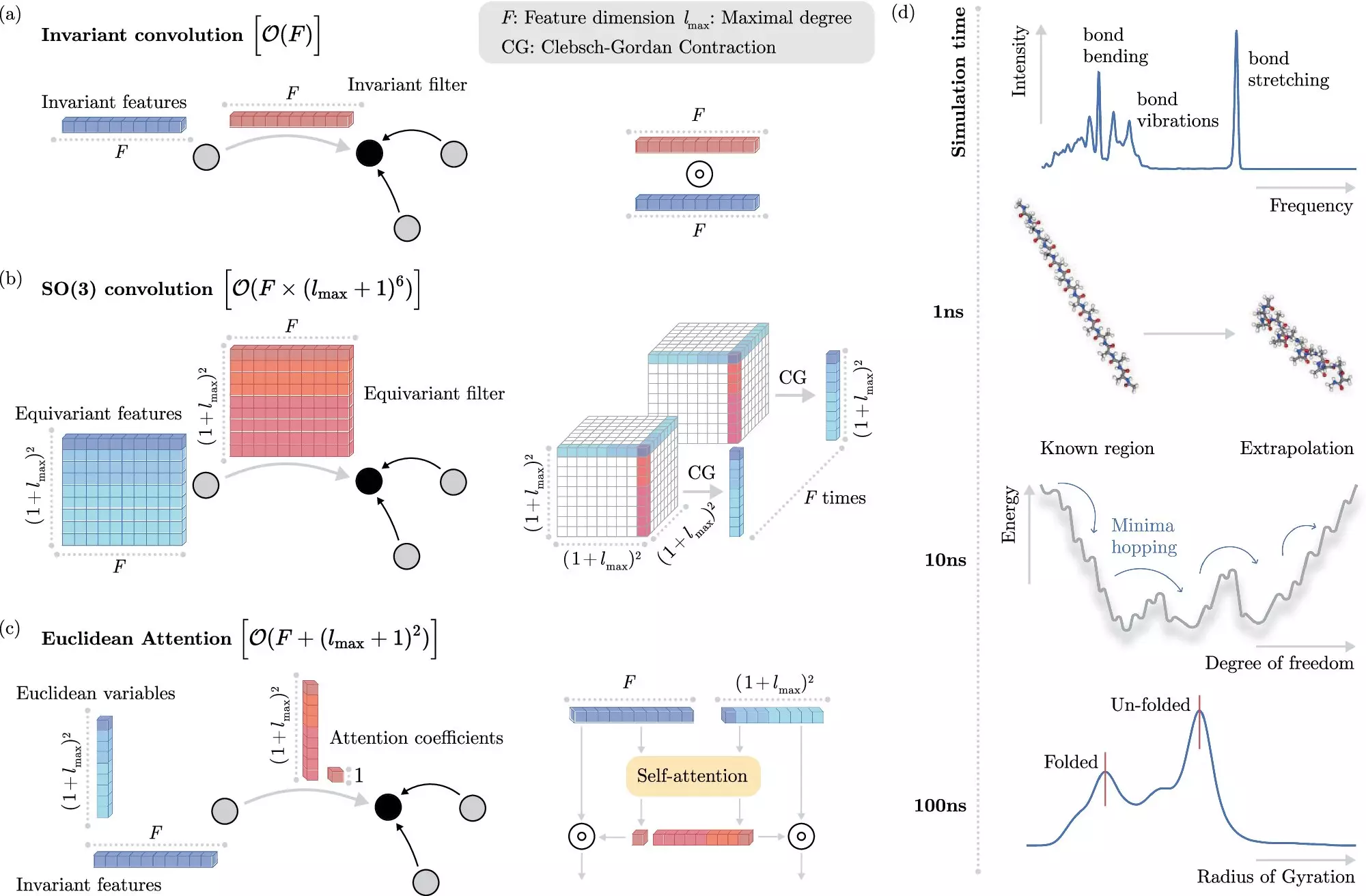
Understanding the behavior of molecules is crucial in various fields such as drug development, material design, and computational chemistry. The simulation of molecular dynamics poses a significant challenge due to the complex interactions between electrons in atoms. Traditional methods rely on solving the Schrödinger equation, which can be computationally expensive and time-consuming. However, recent advancements in machine learning have paved the way for more efficient simulations with reduced computational costs.
Machine learning algorithms have revolutionized the field of molecular dynamics simulations by enabling researchers to predict electronic interactions at the atomistic level without explicitly solving the Schrödinger equation. These algorithms leverage invariances in physical systems to simplify the learning process. By decoupling invariances from other information about chemical systems, researchers can significantly reduce the computational cost of simulations, allowing for faster and more accurate results.
Researchers from the Berlin Institute for the Foundations of Learning and Data (BIFOLD) at TU Berlin and Google DeepMind have developed a novel machine learning algorithm that revolutionizes molecular dynamics simulations. This new approach simplifies the process by allowing the ML model to focus on the most critical operations for predicting electronic interactions accurately. As a result, long-time scale simulations that previously required months or even years of computation can now be performed within a few days on a single computer node.
The implications of this breakthrough are far-reaching, with potential applications in drug development, material design, and computational chemistry. Simulating the interaction of molecules with proteins in the human body could lead to the development of new drugs without the need for costly and time-consuming experiments. Additionally, the ability to identify stable molecular structures with high accuracy opens up new possibilities for research in various fields.
The future of molecular dynamics simulations lies in developing algorithms that can accurately simulate complex, long-range physical interactions in large systems. By combining advanced machine learning techniques with physical principles, researchers can overcome longstanding challenges in computational chemistry and pave the way for more efficient and environmentally friendly scientific research.
Overall, the breakthrough in molecular dynamics simulations represents a significant leap forward in our understanding of atomistic systems. By harnessing the power of machine learning, researchers can unlock new insights into the structure, dynamics, and functioning of molecules, leading to groundbreaking discoveries in various scientific disciplines.
Leave a Reply